Une méthode de calcul plus efficace pour explorer la réactivité des matériaux
Des scientifiques du LAAS-CNRS
Pour mieux comprendre et prédire les propriétés macroscopiques des matériaux, les spécialistes de la modélisation réalisent des calculs à l'échelle atomique, qui doivent être étendus à des systèmes comprenant des centaines ou des milliers d'atomes. Le calcul des chemins de diffusion des atomes permet en particulier d'explorer la réactivité du matériau. Mais les méthodes utilisées jusqu'à maintenant pour construire ces modèles ont l'inconvénient, faute d'être suffisamment rapides, d'exiger des ressources informatiques importantes. Des scientifiques du LAAS-CNRS ont trouvé un moyen d'accélérer et d’améliorer considérablement ces calculs. Ainsi, en appliquant leur nouvelle méthode à l'étude de la diffusion des défauts atomiques dans le silicium, l'équipe a montré que les calculs pouvaient s'effectuer jusqu'à 10 fois plus vite que par des méthodes traditionnelles.
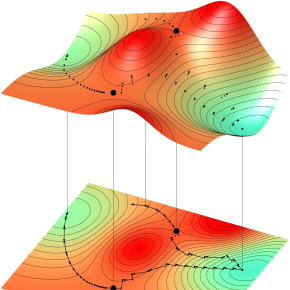
Le calcul des chemins de diffusion atomique est fondé sur l'exploration des surfaces d'énergie potentielle, qui représentent les variations de l'énergie totale du système atomique, les "creux" et les "pics" figurant les minima et maxima d'énergie. L'enjeu est de prédire les chemins possibles entre deux minima d'énergie. Les algorithmes développés au LAAS permettent de calculer efficacement un état final à partir d'un minimum connu, d'identifier exactement un état de transition entre deux minima connus, ou encore de reconstruire un chemin complexe entre deux états donnés.
La nouvelle méthode repose sur un couplage optimisé entre deux techniques de calcul: la théorie fonctionnelle de la densité (DFT, pour Density functional theory), pour le calcul de la structure électronique du matériau, et la technique d'activation relaxation (ART), première méthode numérique, mise au point en 1996, capable de trouver des chemins de transition pour de grands systèmes d'atomes.
Après avoir validé le couplage DFT-ART sur la diffusion des défauts dans le silicium, phénomène qui a un impact sur les propriétés des composants électroniques, l'équipe du LAAS explore d'autres applications potentielles. La nouvelle méthode de calcul est ainsi appliquée aux mécanismes de croissance des couches d'interface dans les semiconducteurs, et à la détection de gaz par des capteurs solides. D'autres utilisations sont possibles, par exemple pour l'étude du stockage de l'hydrogène, ou encore dans le domaine de la catalyse.
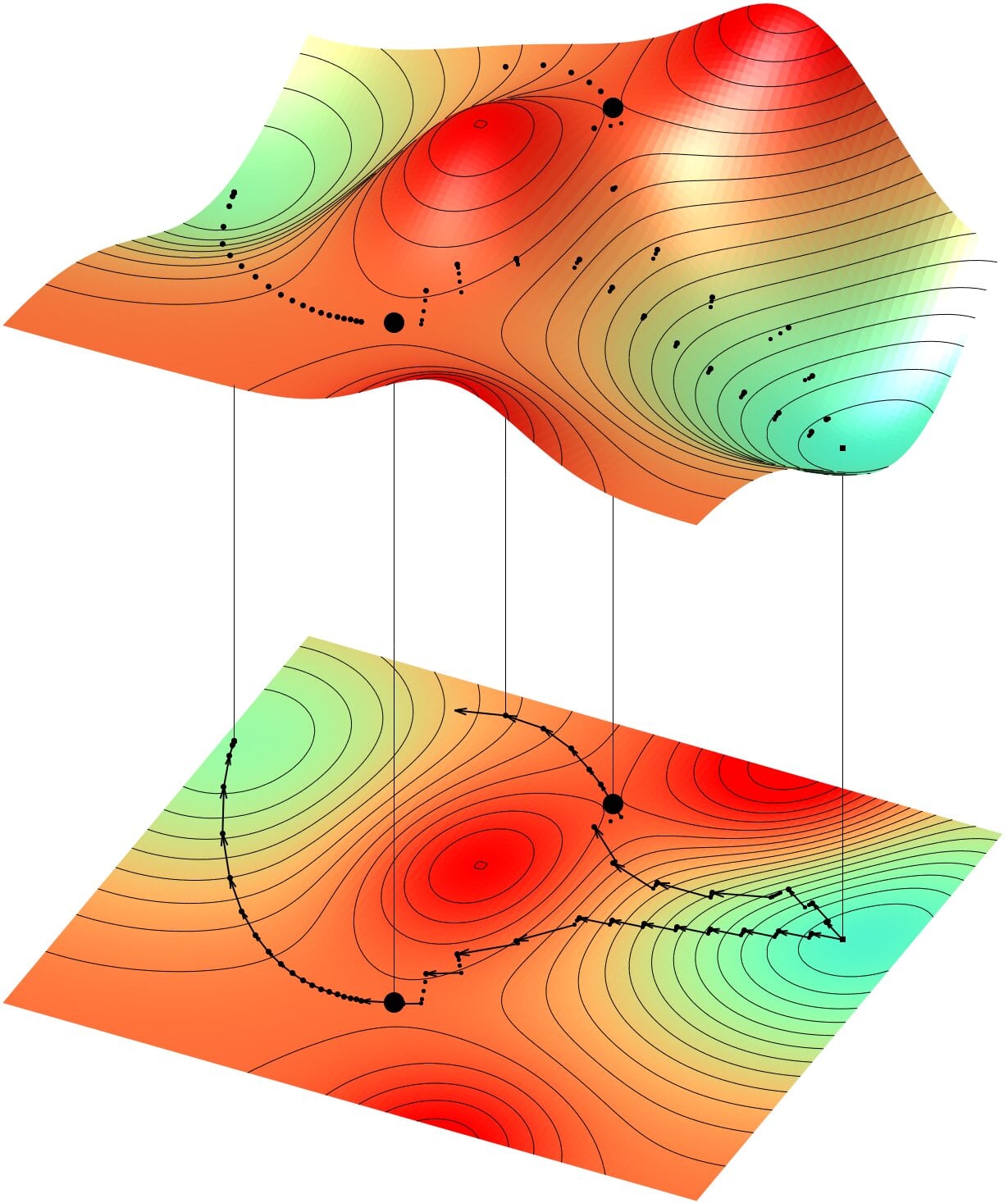
© Antoine JAY, Anne HEMERYCK, LAAS-CNRS
Références
Finding Reaction Pathways and Transition States: r‑ARTn and d‑ARTn as an Efficient and Versatile Alternative to String Approaches
A. Jay, C. Huet, N. Salles, M. Gunde, L. Martin-Samos, N. Richard, G. Landa, V. Goiffon, S. De Gironcoli, A. Hémeryck, N. Mousseau
J. Chem. Theory Comput. 2020, 16, 6726−6734
https://dx.doi.org/10.1021/acs.jctc.0c00541